
Plenary Lecture
Kinetics of Structural Transformations in Nano-Structured Intermetallics: Atomistic Simulations

Professor Rafal Kozubski
Interdisciplinary Centre for Materials Modelling
M. Smoluchowski Institute of Physics
Jagiellonian University in Krakow
Poland
E-mail: rafal.kozubski@uj.edu.pl
Abstract: Kinetics of vacancy-mediated atomic ordering processes in nano-layered L10 and triple-defect B2 ordered intermetallics was the subject of extensive atomistic simulations. The two groups of systems differ substantially in their vacancy thermodynamics: very low and very high vacancy concentration is observed in L10 and triple-defect B2 intermetallics, respectively. Special attention was focused on the analysis of an effect of free surfaces on superstructure stability and defect concentration in the examined materials.
Two models of L10-ordered FePt intermetallic: Ising-type model with two-body interactions and a model with many-body interactions based on Analytic Bond-Order Potentials (ABOP) were simulated by the Quasi-Kinetic Monte Carlo (q-KMC) technique implemented with the vacancy mechanism of atomic migration. For the ABOP model the method was combined with Molecular Statics (MS). Simulation of “order-order” kinetics in [001]-oriented FePt nanolayers initially perfectly ordered in the c-variant L10 and modelled with two-body interactions revealed a tendency for superstructure transformation from c-variant (monoatomic planes parallel to the (001) free surface) to a- and b-variants (monoatomic planes perpendicular to the (001) free surface) (Fig.1) [1]. The transformation showed complex kinetics which, except for uniform (bulk-like) disordering, involved three processes: (i) nucleation of a- and b-variant L10 domains at the surface being initially a single atomic Fe layer, (ii) slow fluctuating growth of the nucleated a- and b-variant L10 domains inward the layer and (iii) relaxation of the microstructure of the surface domains. In sufficiently thin layers, a percolation of the a- or b-variant superstructure domain nucleated at the surface through the layered sample was observed.
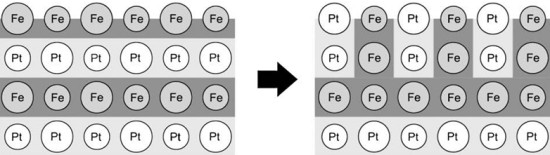
Fig.1. Scheme of an initial stage of L10 c-variant/a(b)-variant transformation in a FePt layer (atoms residing on two nn crystallographic planes are represented by small and big circles) [1].
MC simulations of ABOP-based model of the same L10 FePt layers revealed strong attraction of vacancies by free surfaces. Complex atomic ordering kinetics was observed. Initially fast partial disordering of an internal part of a layer was followed by surface disordering with no L10 c-variant to a(b)-variant transformation. The transformation, however, was observed in additional Monte Carlo/Static Relaxation simulations performed with the direct exchange algorithm [2,3].
The configurational energy of (001)-oriented FePt layers L10-ordered in c- and a(b)-variants was calculated with (i) an Ising-type model, (ii) an ABOP model and (iii) a DFT-based model. In all cases, a- and b-variants of L10 resulted in being energetically stable.
Remarkably, the L10 c-variant -> a(b)-vasriant transformation was experimentally observed in FePt epitaxially deposited multilayers [4].
Triple-defect formation in B2-ordered NiAl intermetallic compound results from a strong asymmetry between the formation energies of Ni- and Al-antisite defects. Chemical disordering in the system is strictly correlated with vacancy formation, which is the reason for the very high vacancy concentration. As a consequence, Kinetic Monte Carlo (KMC) simulation of ordering occurring in the system and controlled by atomic migration via the vacancy mechanism must involve complete vacancy thermodynamics – i.e. the simulated system must contain an equilibrium concentration of vacancies. NiAl was modelled with an Ising-type Hamiltonian and the temperature-dependent equilibrium concentration of vacancies was determined by means of Semi Grand Canonical Monte Carlo (SGCMC) simulations [5], which assured consistency of the entire approach. The SGCMC simulations led to the evaluation of nearest-neighbour (nn) pair-interaction energies generating the triple-defect behavior of the system. The system generated and modelled in the same way as in the SGCMC was then simulated by KMC for “order-order” kinetics. The procedure required in addition the determination of saddle-point energies assigned to particular atomic jumps to nn vacancies. Their values were estimated in relation to the nn pair-interaction energies with reference to MS simulations performed for NiAl with embedded atom method (EAM) energetics.
The procedure was comparatively applied to bulk and nano-layered B2 AB systems. In both cases, the KMC simulations were started from a configuration with no antisite defects and vacancies (whose number resulted from SGCMC) distributed at random. The results elucidated the role of triple-defect formation as the atomistic-scale origin of the experimentally observed (surprising) low rate of “order-order” kinetics in bulk NiAl. The simulated “order-order” kinetics showed two stages: (i) extremely fast generation of triple defects – i.e. creation af A-antisite defects and related shift of almost all B-vacancies to A-sublattice; the process, which, however, did not lead the system to thermodynamic equilibrium, (ii) extremely slow continuation of the process towards thermodynamic equilibrium – i.e. equilibrium concentration and configuration of antiside defects and vacancies (Fig.2).

Fig.2. KMC simulated “order-order” kinetics in B2-ordering triple-defect binary AB system: ηA is a Bragg-Williams-type long-range order parameter [6,7].
It was shown that the slow rate of the stage (ii) was due to extremely low efficiency of disordering jumps of A-atoms, which were reversed with very high probability resulting from numerous vacancies residing on A-sublattice. It is claimed that only the stage (ii) of “order-order” kinetics is observed experimentally.
In nano-layers, an additional effect of vacancy segregation on free surfaces and its influence on ordering kinetics was modelled and compared with the related Molecular Dynamics results [8].
[1] M. KOZŁOWSKI, R. KOZUBSKI, CH. GOYHENEX, V. PIERRON-BOHNES, M. RENNHOFER AND S. MALINOV, Atomic Ordering in Nano-Layered FePt, Intermetallics 17 (2009) 907-913.
[2] R. KOZUBSKI, M. KOZLOWSKI, J. WROBEL, T. WEJRZANOWSKI, K. J. KURZYDLOWSKI, CH. GOYHENEX, V. PIERRON-BOHNES, M. RENNHOFER AND S. MALINOV, Atomic Ordering in Nano-layered FePt: Multiscale Monte Carlo Simulation , Comput. Mater. Sci. 49 (2010) S80-S84.
[3] M. KOZLOWSKI, Thesis, Jagiellonian University in Krakow, 2010
[4] M. RENNHOFER, M. KOZLOWSKI, B. LAENENS, B. SEPIOL, R. KOZUBSKI, D. SMEETS AND A. VANTOMME, Study of reorientation processes in L10-ordered FePt thin films, Intermetallics, 18 (2010) 2069-2076.
[5] A. BIBORSKI, L.ZOSIAK, R. KOZUBSKI, R.SOT AND V. PIERRON-BOHNES, Semi-Grand Canonical Monte Carlo simulation of ternary bcc lattice-gas decomposition: Vacancy formation correlated with B2 atomic ordering in A-B intermetallics, Intermetallics, 18 (2010) 2343-2352.
[6] A. BIBORSKI, Thesis, Jagiellonian University in Krakow/Université de Strasbourg, 2010.
[7] P. Sowa, R. Kozubski, A. Biborski, E.V. Levchenko, A.V. Evteev, I. V. Belova, G.E. Murch, V. Pierron-Bohnes, “Self diffusion and “order-order” kinetics in B2-ordering AB binary systems with a tendency for triple defect formation: Monte Carlo simulation”, Philos.Mag. (2012) doi:10.1080/14786435.2012.742591
[8] E.V. LEVCHENKO, A.V. EVTEEV, R. KOZUBSKI, I. V. BELOVA AND G.E. MURCH, Molecular Dynamics Simulation of Surface Segregation in a (110) B2-NiAl Thin Film, Phys. Chem. Chem. Phys., 13 (2011) 1214-1221.
Brief Biography of the Speaker: Prof. Rafal Kozubski academic carrier: 1984 Ph.D., Jagiellonian University in Kraków, 1987 - 1988 post-doctoral position, Strasbourg Institute of Physics and Chemistry of Materials (IPCMS), France, 1988, 1990, academic visitor, Institute for Applied Physics, Swiss Federal Institute of Technology, Zurich, Switzerland, 1993 - 1995 Lise-Meitner Fellow, Institute for Solid State Physics, University of Vienna, Austria, 1997 habilitation (DSc), Jagiellonian University in Kraków, 1997 - 2006 associate professor, 2006 - full professor in the Jagiellonian University, Kraków, 2006-2008 International Fellow, Queen’s University in Belfast, 2007, 2008, 2009, 2010, 2011, 2012, 2013 Visiting professor L.Pasteur University in Strasbourg/University of Strasbourg. Research output: over 100 publications in international reviewed journals, over 140 communications on international conferences.
https://www.usosweb.uj.edu.pl/kontroler.php?_action=actionx:katalog2/osoby/pokazOsobe(os_id:51027)&lang=2